Seleccione un país o región


-
Argentina (Español)

-
Australia (English)

-
Austria (Deutsch)

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil (Portugues)

-
Canada (English)

-
Canada (Français)
-
Chile (Español)

-
Colombia (Español)

-
Croatia (Croatian)

-
Denmark (Danish)

-
Deutschland (Deutsch)

-
Europe (English)

-
France (Français)

-
Greece (Ελληνικά)

-
Italia (Italiano)

-
Hungary (Magyar)

-
Lietuva (Lietuviškai)

-
Mexico (Español)

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland (Nederlands)

-
Norge (Norsk)

-
Oman (العربية)

-
Oman (English)
-
Polska (Polskie)

-
Portugal (Portuguese)

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain (Español)
-
Suomi (Suomi)

-
Sverige (Svenska)

-
Schweiz (Deutsch)

-
台灣 (中文)

-
United States (English)

-
UAE (العربية)

-
UAE (English)
Las personas que aparecen en la imagen son pacientes reales que han dado su consentimiento, bien directamente o a través de sus familias, para utilizar sus historias. Las imágenes se incluyen meramente a título ilustrativo


¿QUÉ ES LA AME?
¿QUÉ CAUSA LA AME?
La atrofia muscular espinal (AME) es una enfermedad neuromuscular genética rara provocada por una mutación en el gen de supervivencia de las motoneuronas 1 (SMN1).1


¿QUÉ ES LA AME?
TIPOS DE AME
Existen varios tipos de AME en función de la edad de inicio y la capacidad funcional. Además, dentro de cada tipo, hay diversos grados de gravedad.2


¿QUÉ ES LA AME?
¿CÓMO SE HEREDA LA AME?
LA AME es una enfermedad hereditaria cuya causa genética específica, a diferencia de otros muchos trastornos neuromusculares, es bien conocida.3
¿QUÉ ES LA AME?
ENFERMEDADES CON SÍNTOMAS SIMILARES
Aunque la AME se caracteriza por determinados signos y síntomas, hay otras enfermedades que tienen síntomas similares pero con causas genéticas diferentes.

¿QUÉ ES LA AME?
DIAGNÓSTICO Y PRUEBAS
Cuando se sospecha una AME, es importante confirmar el diagnóstico inicial con un análisis genético.4
En Biogen, estamos comprometidos a apoyar a aquellas personas con atrofia muscular espinal y al equipo encargado de su cuidado. Nuestra esperanza para Unidos en la lucha contra la AME es que, proporcionando la información necesaria, podamos ayudarle a conocer la AME.



¿QUÉ ES LA AME?
¿QUÉ CAUSA LA AME?
La atrofia muscular espinal (AME) es una enfermedad neuromuscular genética rara provocada por una mutación en el gen de supervivencia de las motoneuronas 1 (SMN1).1

¿QUÉ ES LA AME?
TIPOS DE AME
Existen varios tipos de AME en función de la edad de inicio y la capacidad funcional. Además, dentro de cada tipo, hay diversos grados de gravedad.2

¿QUÉ ES LA AME?
¿CÓMO SE HEREDA LA AME?
LA AME es una enfermedad hereditaria cuya causa genética específica, a diferencia de otros muchos trastornos neuromusculares, es bien conocida.3
¿QUÉ ES LA AME?
ENFERMEDADES CON SÍNTOMAS SIMILARES
Aunque la AME se caracteriza por determinados signos y síntomas, hay otras enfermedades que tienen síntomas similares pero con causas genéticas diferentes.
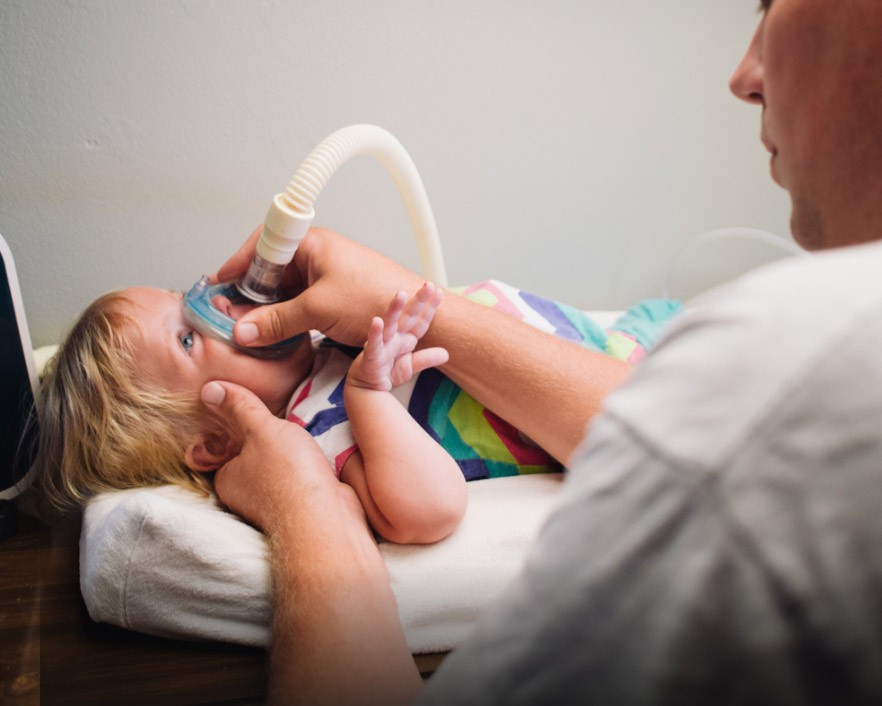
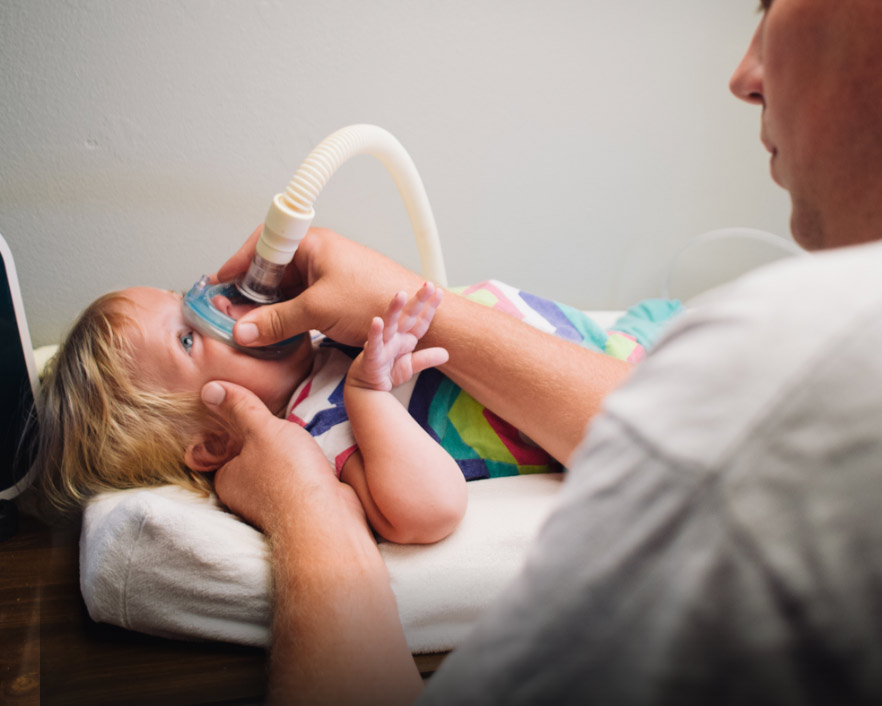
¿QUÉ ES LA AME?
DIAGNÓSTICO Y PRUEBAS
Cuando se sospecha una AME, es importante confirmar el diagnóstico inicial con un análisis genético.4
En Biogen,estamos comprometidos a apoyar a aquellas personas con atrofia muscular espinal y al equipo encargado de su cuidado. Nuestra esperanza para Unidos en la lucha contra la AME es que, proporcionando la información necesaria, podamos ayudarle a conocer la AME.
Las personas que aparecen en la imagen son pacientes reales que han dado su consentimiento, bien directamente o a través de sus familias, para utilizar sus historias.
Las imágenes se incluyen meramente a título ilustrativo.
“El proceso de diagnóstico de la AME no ha cambiado desde el artículo original sobre la declaración de consenso... a menos que haya casos familiares previos, el proceso de diagnóstico se inicia con la sintomatología clínica.” 4
Bibliografía
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
2. Kolb SJ, Kissel JT. Spinal muscular atrophy. Arch Neurol. 2011;68(8):979-984.
3. National Organization for Rare Disorders. Spinal Muscular Atrophy. [online] 2012 [cited 2020 Sep 22].
Available from: URL: https://rarediseases.org/rare-diseases/spinal-muscular-atrophy/.
4. EGL Genetics. Congenital Hypotonia Panel: Spinal Muscular Atrophy Deletions, Prader-Willi/Angelman Syndrome Methylation, Myotonic Dystrophy, and Uniparental Disomy 14. Available at: http://geneticslab.emory.edu/tests/HY. Accessed January 9, 2017.
5. Mercuri E, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018;28(2):103-115.