Seleccione un país o región


-
Argentina (Español)

-
Australia (English)

-
Austria (Deutsch)

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil (Portugues)

-
Canada (English)

-
Canada (Français)
-
Chile (Español)

-
Colombia (Español)

-
Croatia (Croatian)

-
Denmark (Danish)

-
Deutschland (Deutsch)

-
Europe (English)

-
France (Français)

-
Greece (Ελληνικά)

-
Italia (Italiano)

-
Hungary (Magyar)

-
Lietuva (Lietuviškai)

-
Mexico (Español)

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland (Nederlands)

-
Norge (Norsk)

-
Oman (العربية)

-
Oman (English)
-
Polska (Polskie)

-
Portugal (Portuguese)

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain (Español)
-
Suomi (Suomi)

-
Sverige (Svenska)

-
Schweiz (Deutsch)

-
台灣 (中文)

-
United States (English)

-
UAE (العربية)

-
UAE (English)
¿QUÉ ES LA ATROFIA MUSCULAR ESPINAL (AME)?
La AME afecta a la parte del sistema nervioso que controla el movimiento muscular voluntario.1 En la atrofia muscular espinal se produce una pérdida de células importantes de la médula espinal, denominadas motoneuronas, que son fundamentales para la fuerza y el movimiento muscular. Estas motoneuronas regulan la actividad muscular enviando señales desde el sistema nervioso central (SNC), que es la parte del sistema nervioso que incluye el cerebro y la médula espinal.1,2
Cuando el músculo deja de recibir señales del SNC, la pérdida de motoneuronas funcionales provoca una debilidad y una atrofia muscular progresivas (la disminución gradual de la masa y la fuerza muscular).3
CLAVES PARA ENTENDER LA CAUSA DE LA ATROFIA MUSCULAR ESPINAL
A diferencia de muchas otras enfermedades neuromusculares raras, en el caso de la atrofia muscular espinal se conoce bien la causa genética específica. La causa de la AME es una mutación en el gen de supervivencia de las motoneuronas 1 (SMN1), que es el responsable de producir la proteína de supervivencia de las motoneuronas (SMN). Esta proteína es la encargada de mantener el buen estado y el funcionamiento normal de las motoneuronas.
En las personas con atrofia muscular espinal, las dos copias del gen SMN1 sufren una mutación que provoca una reducción en la producción de la proteína SMN. Sin los niveles adecuados de proteína SMN, se pierden las motoneuronas de la médula espinal, por lo que los músculos no reciben las señales apropiadas del cerebro.4
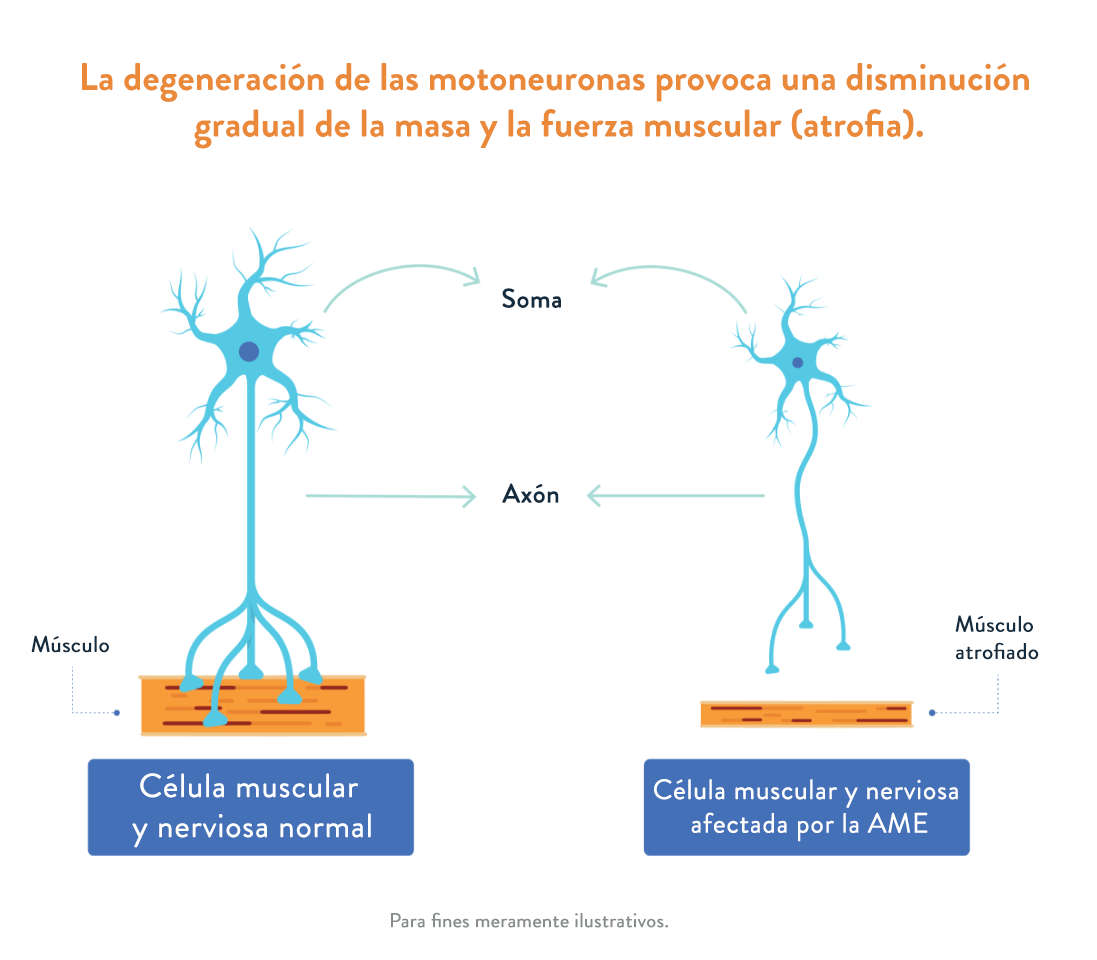
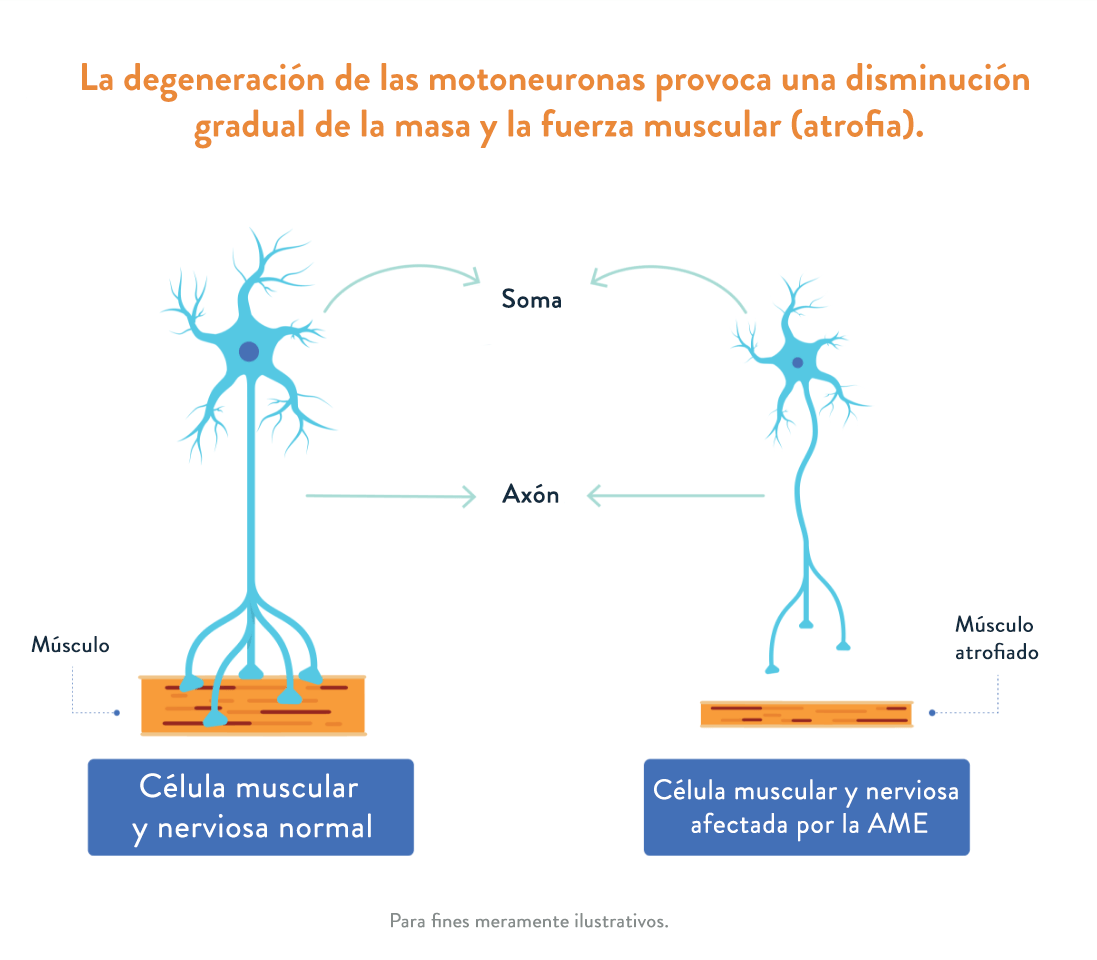
Existen varios tipos de AME en función de la edad de inicio y la capacidad funcional. Además, dentro de cada tipo hay diversos grados de gravedad.5


CONÉCTESE
para más información sobre cómo formar parte de la comunidad AME
Las personas que aparecen en la imagen son pacientes reales que han dado su consentimiento, bien directamente o a través de sus familias, para utilizar sus historias. Las imágenes se incluyen meramente a título ilustrativo.
Bibliografía
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
2.Wang CH, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-1049.
3. National Institute of Neurological Disorders and Stroke. Motor Neuron Disease Fact Sheet. 2012.
Available at: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Motor-Neuron-Diseases-Fact-Sheet. Accessed January 9, 2017.
4. Genetics Home Reference. SMN2 gene. 2012. Available at: https://ghr.nlm.nih.gov/gene/SMN2. Accessed January 9, 2017.
5. Kolb SJ, Kissel JT. Spinal muscular atrophy. Arch Neurol. 2011;68(8):979-984.